
Predicting protein dynamics, moats and commercialization
Combining technological tailwinds for maximally defensible businesses

Bridging the target and phenotype gap
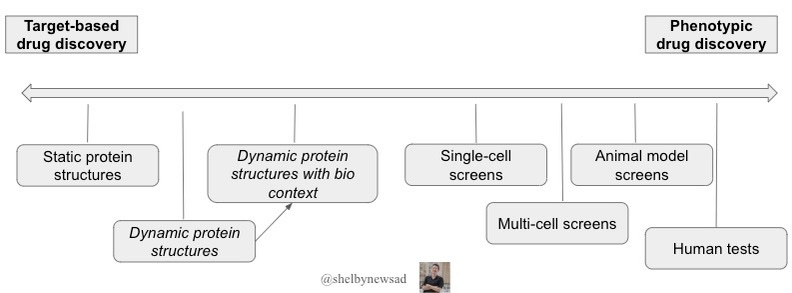
Organisms and their parts are dynamic. Measuring and understanding and then controlling these dynamic systems is the crux of every bio company. There are two ways companies have historically gone about this - either by embracing complexity (phenotypic drug discovery, PDD) or reducing the complexity (target-based drug discovery, TDD).
To embrace complexity, there are pushes to capture more data by characterizing cellular phenotypes (Recursion, Insitro, and many smaller companies) and animal phenotypes (Vevo Therapeutics, Psychogenics) in response to treatments. After all, phenotypic drug discovery disproportionately finds first-in-class medicines and more recently has been used to discover new drugs for cystic fibrosis, hepatitis C, and multiple myeloma.
On the other hand, reductionist drug discovery (called target-based drug discovery; TDD) is still the most common. New protein folding models are typically applied to target-based drug discovery because of the structure and binding of drugs like it. The issue with this mode of drug discovery - or even solving protein structures (with X-ray crystallography or Cryo-EM) and seeing how drugs bind is that these are static structures. Even the newest models such as Chai-11 and Boltz-1, predict static protein structures. However, proteins are naturally flexible with many different shapes possible natively and through post-translational modifications.
What I’m proposing is that researchers and companies will start bridge the TDD and PDD gap by more deeply understanding diseased state proteins (splicing and chemical modifications) and using this information for more predictive computational modeling.
Increased biological context
Recently, increasing amounts of biological context of proteins themselves has been unlocked. The real workhorse of proteomics is mass spectrometry which can identify 8000+ proteins, the proteins’ splicing variants and post-translational modifications. Continuous innovation building on top of data-independent acquisition (DIA) such new labeling techniques have led to advancements which mean we can now do proteomics on single-cells. Understanding protein dynamics in living cells has importantly been an extension of mass spectroscopy which is directly useful to better structure predictions, helping to eventually close the gap between TDD and PDD. Other nascent technologies such as Nanopore proteomics will be helpful especially to advance single-cell and non-biased post-translational modifications readouts in proteomics2. Several companies are working on Nanopore and fluorescent-based proteomics (Glyphic, Nautilus, Erisyon, QSi, Portal).
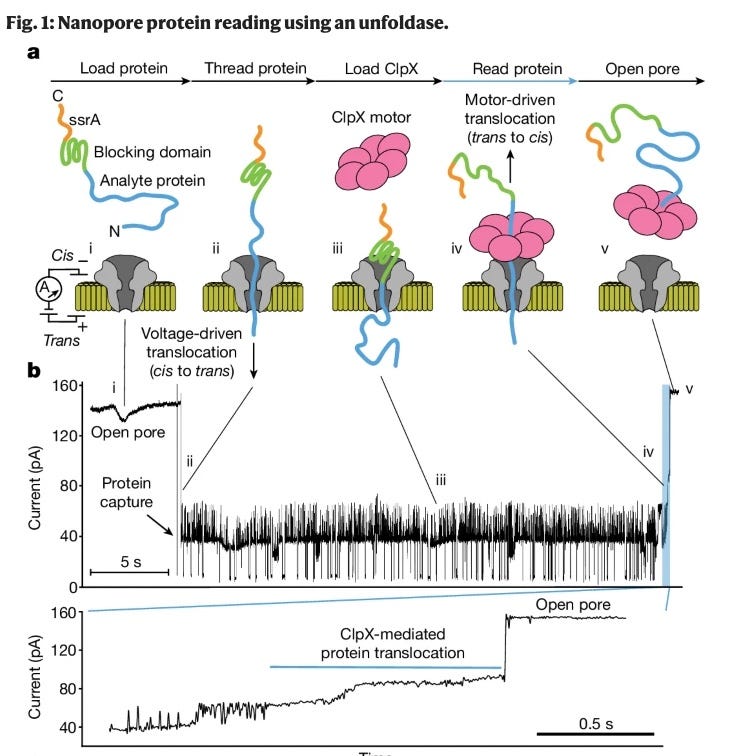
Graphic of nanopore-based sequencing technology and data readouts.
Though the importance of increased biological context data is a fairly consensus view even by the most computational teams (with Evolutionary Scale hiring wet lab scientists), we’ve heard fewer people speaking specifically about *which* data exactly will add the most value which is why we think it’s important to explicitly talk about advances in proteomics.
Model development around protein conformation prediction
Having increased biological context is only part of the solution, the other part is predicting how the biological changes influence the protein structure itself. The computational prediction of protein conformations has correspondingly been making many advances around three central premises:
(1) Improving sub-sampling protein sequence inputs
(2) Innovations building on molecular dynamics
(3) Generative models using PDB training with/out MD
Today, using a combination of these methods and doing comparative Boltzman rankings are the way to best understand likely conformations, if no additional biological context is added. In the future we see physics-based and generative models (and combination thereof) being the technology areas with highest chance of step change breakthroughs.
1. From sequence sub-sampling
Some work has been completed on sub-sampling of multiple sequence alignments (MSA) of proteins. What this means is that MSA inputs are subsampled OR mutated (in the case of SEARCH_AF), and then run through AlphaFold2. This works because if the input MSA’s change, the coevolutionary network changes. This is important because Alphafold2 is built on coevolution because amino acid changes which physically interact with other amino acids to create compensatory mutations to ensure continued protein function. Coordinated changes seen in MSAs create a picture of what residues are interacting and can be coordinated to predict (with sub-sampling) diverse protein conformational ensembles. Again, this is very helpful today but don’t see the next step-change being in this space. This is due to an inability for MSA models to be applied to the popular protein language models and protein databank (PDB) molecular dynamic ensembles can’t be incorporated3.
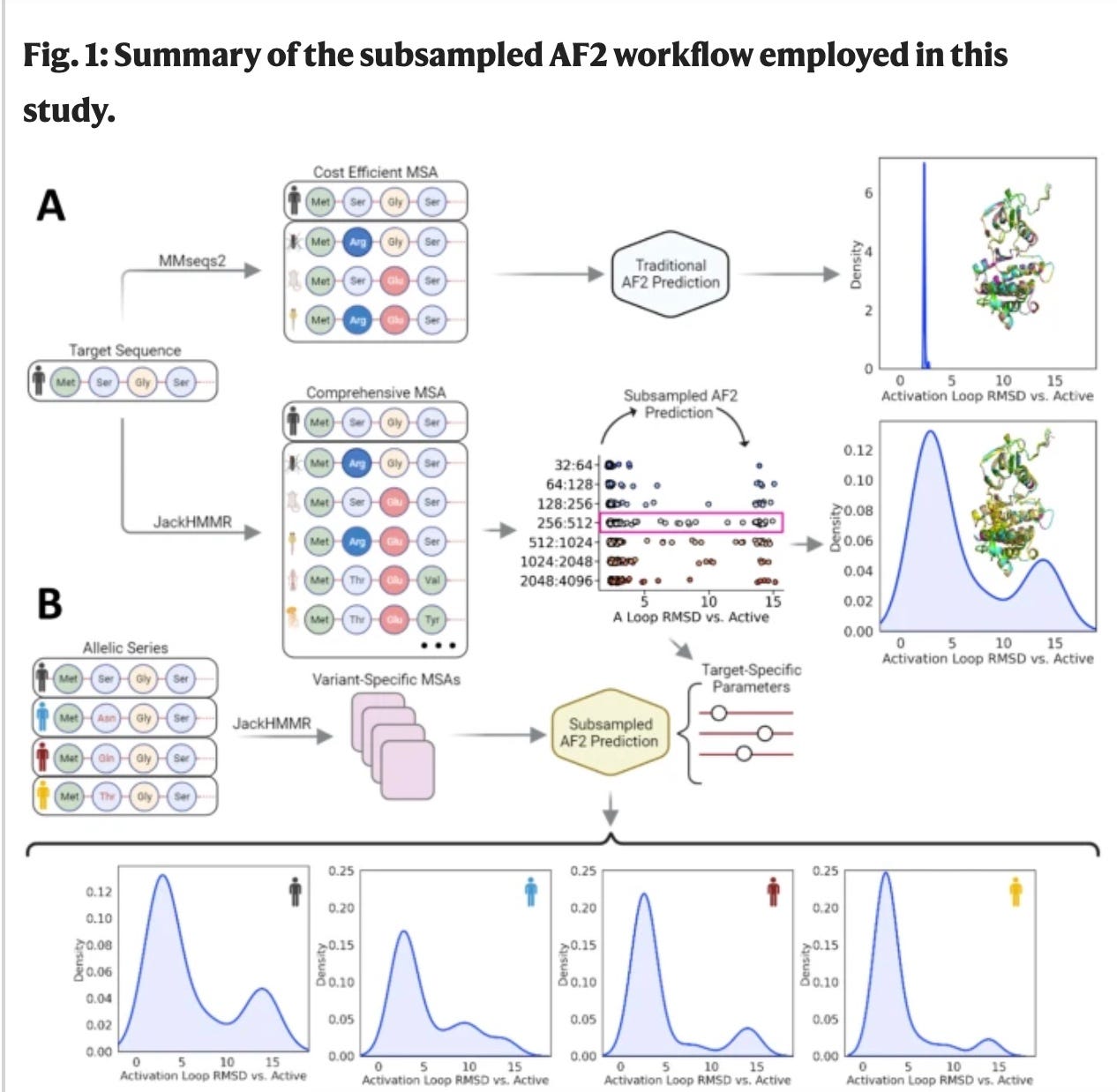
2. Building on molecular dynamics
MD predictions are useful as their core capability is to understand protein states over time (which is much more valuable than small snapshots). Due to understanding the protein over time, MD simulations can capture metastable states and rare conformations. The added benefits are that protein states over time can be modeled in different cellular environments (cell membrane vs. internal and acid vs. basic conditions) to create a more holistic picture of protein states. Unfortunately, MD simulations are computationally intensive and slow. Since their initial publication with Alder and Wainwright in 1957 scientists have been working to improve the timescales, computational efficacy, and size of systems MD can predict. Methods to improve efficacy include Markov state models (model probability of transitions between different conformational space), enhanced subsampling (umbrella sampling and metadynamics), coarse grained models (with force fields, top-down predictions with diffusion models and bottom up from all-atom simulations), and neural net potentials (NNPs; discussed at length here by my colleague Mackenzie).
We’ve also been encouraged by initiatives such as the Dynamic Protein Data Bank which has aggregated molecular dynamic simulations of >12,000 proteins and then added context to the conformational path by adding diffusion models.
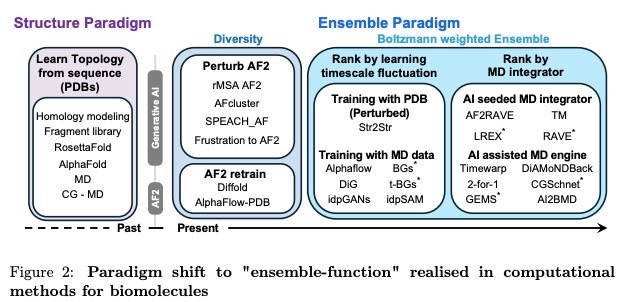
3. Generative models using PDB training with/out MD
Many of the ML models in the protein conformation space aren’t trained on MD simulations, they’re trained on the static structures of the Protein Data Bank (PDB). AlphaFold3 follows this trend, using a diffusion approach for the sampling of different protein structures as does the ability to predict protein conformation while interacting with other biomolecules (DNA/RNA, proteins, small molecules). Despite the diversity of structures it can generate, it still doesn’t predict pathways of conformations nor likelihoods of one structure vs. another which is important information for making drugs. There’s a surge of technology which are using generative models trained on the PDB (some being enhanced by MD simulations and MSAs):
Str2Str uses an SE(3) diffusion architecture to iteratively perturb the initial structure of a protein’s backbone to get an ensemble of conformations with training on the PDB alone.
EigenFold is also trained on the PDB and uses harmonic diffusion to constrain conformational outputs to those physically possible.
Distributional Graphformer (DiG) uses PDB structures, MSAs and MD simulations data with a graph neural network architecture to predict molecular conformations in equilibrium.
AlphaFLOW uses a diffusion-like process called flow matching to sample conformational states after being trained on PDB and MD simulation data.
Emerging frameworks for commercialization
The vision of computational biology is that one day we will be able to have zero-shot drug design based on complete conformational modeling with biological context. Those which traverse this path can start to bridge TDD and PDD drug discovery which will lead to more efficacious drug discovery.
Companies that want to build in this arena have the difficult job of pairing technology that adds biological context, with computational models, and (likely) asset furthering excellence. The goal isn’t just to have a moat/unfair advantage/wedge today, it’s to build in perpetual dominance. I once saw a meme comparing the Warren Buffet idea of moats to those of Jeff Bezos and think it's fitting in the era where there’s data scarcity around deep biological understanding of many potential targets and noise due to how many new models are being published4. Complicating things, there’s the very real bottleneck of needing the drugs made from this new system still need to go through human trials.

Commercially, there’s a mismatch between the academic output of papers predicting protein conformations and number of startups (Superluminal and Allotec are only companies we’ve seen centered around protein dynamics). We see this mismatch as an opportunity for where drug discovery has to go. To reiterate, where we see the highest chance of success is combining technological tailwinds (proteome analysis + SOTA computation) to make better assets. There is nuance here if the founding team has business development excellence and has been able to sell into top players without asset furthering (though this is more exception than the rule) and deciding requires a deeply individual assessment.
At a high level, a framework to maximize impact in drug discovery is that founders should build the hardest version of the company as possible. Building every business is hard which is why building the maximally ambitious and defensible version makes the pursuit worth founders’ time.
We’re enthused to speak with folks thinking in similar ways - do reach out if this piece resonates.
_________________________________________________
Thanks to Mike Dempsey, Collin Spencer and Sidharth Sirdeshmukh for comments on this piece.
Chai-1 does allow for prompting with new data (such as lab restraints) which improves prediction performance.
Many companies working here on Nanopore and fluorescence-based measures (Glyphic, Nautilus, Erisyon, QSi, Portal).
Not to mention that models have fleeting moats.
also see counter-arguments: https://www.quantamagazine.org/how-ai-revolutionized-protein-science-but-didnt-end-it-20240626/
Great post! Wondering if you think there will be any large changes due to new understandings (perhaps most important for molecular dynamics) that arise from application of non-linear statistical methods.
Of course part of the reductionist approach is over reliance on traditional linear statistical methods, yet biology is inherently nonlinear.
Then, say we do get new insights. Does this lead us back to a single (or very limited number of) drug targets and then standard drug development?